Computational Chemistry
The Computational Chemistry research group is led by Prof. Kari Laasonen and is currently the home of three postdoctoral researchers and three doctoral students. A short presentation of the group, our ongoing research activities and a list of recent publications can be found here. The current status of job openings in the research group including Master's thesis projects and summer employee positions is announced at the bottom.
Computational modelling has emerged as an important research tool in the fields of chemistry and materials design, because it allows us to understand chemical phenomena such as catalysis at the fundamental level of atoms, electrons and molecules. In the future, the importance of computational modelling will continue to grow as increases in computational capacity will enable us to tackle larger and more complex problems. To meet these challenges, there are currently three research groups at the Department of Chemistry and Materials Science who are actively applying and developing new computational methods to study different problems in chemistry. In addition to the Computational Chemistry group, these include the Inorganic Materials Modelling group led by Prof. Antti Karttunen and the Soft Materials Modelling group led by Senior Scientist Maria Sammalkorpi.
Our group's research areas include:
-
Molecular modelling of electrochemical reactions
-
Modelling of surfaces, interfaces and nanoparticles
-
Ab initio molecular dynamics
The unifying theme of all the research projects at the Computational Chemistry group is to understand how the electronic structure of atoms and molecules can be used to explain, and ideally predict, the chemical and physical properties of experimentally interesting systems. To investigate such fundamental properties of molecular systems, a quantum mechanical treatment is necessary. Because chemical processes are complex, our tool of choice is most often density functional theory (DFT), both in its time-independent and time-dependent flavors, where the electron density is the basic quantity governing the behavior of atoms. This allows us to simulate systems up to thousands of atoms in size, as well as time-scales beyond the reach of most other electronic structure theories. The mathematical representations of these computational models are nevertheless very demanding and solving them would not be possible without the supercomputing facilities provided by The Finnish IT Center for Science (CSC).
Our work featured on scientific journal covers


Research topics and projects:
Computational Electrochemistry:
Electrochemical devices, such as batteries and fuel cells, are key components in a wide variety of applications including portable electronics and transportation. To fully understand and develop the performance of these devices, an atomistic description of the structure and electron transfer properties of the employed functional materials is required. Nowadays, density functional theory based computational modelling is routinely applied to gain nanoscale information in the rational design of more efficient catalysts or for verifying reaction mechanisms. However, the field of computational electrochemistry and -catalysis is still in its infancy due to the unique technical challenges associated with treating electron transfer reactions at constant electrode potential conditions. Also the importance of solvation and interfacial dynamics remain to be thoroughly elucidated.
In the Computational Chemistry group, we are interested in electrochemical systems which are also relevant from the viewpoint of experimentalists. Examples include the hydrogen and oxygen evolution reactions on prototypical systems such as single-crystal platinum, as well as novel materials like pristine and modified carbon nanostructures and earth-abundant transition metal compounds (e.g. MoS2, Ni2P). We are also very much interested in the more fundamental aspects of interfacial electrochemistry, namely the structure of dynamic electrode-electrolyte interfaces as well as rigorous description charge transfer processes using formalisms such as constrained density functional theory (CDFT). In exploring the dynamics of electrochemical reactions and interfaces, we apply our solid expertise in performing ab initio molecular dynamics simulations.
To achieve a comprehensive understanding of these systems, we actively collaborate with experimental electrochemistry research groups, for example, with the Electrochemical Energy Conversion group lead by Prof. Tanja Kallio and the research group of Dr. Andy Wain at the National Physical Laboratory (NPL, UK).
Research highlights:
Computational Modelling of Atomic Layer Deposition:
Atomic layer deposition (ALD) is a coating technology used to produce highly uniform thin films. Although most of the ALD research is conducted by conventional experiments, small time-scale surface reactions are difficult to probe. Computational quantum chemistry provides a complementary tool to gain refined insight on the surface kinetics. Understanding these surface reactions is essential in the design and optimization of ALD processes.
The key to bridge molecular simulations with real-size processes is kinetic modelling. Homogeneous systems can often be broken down into a few crucial, rate-limiting steps that can be modelled using macroscopic, text-book rate equations. However, heterogeneous systems - e.g. surface reactions - seldom fulfill the underlying assumptions of macroscopic rate equations. Adsorbed molecules often have lateral interactions and reaction energetics are dependent on the surface coverage. Such phenomena can be described by, for example, kinetic Monte Carlo (kMC) simulations where a stochastic algorithm is used to explore the phase space of surface configurations. Transition rates between different configurational states can be calculated ab initio using density functional theory or post-Hartree-Fock theories.
Research highlights:
First-Principles Thermodynamic Study of Alloys:
Alloys of e.g. the platinum group metals (PGM) have a wide variety of existing and potential applications in catalysis. However, much of the thermodynamic properties of platinoid alloys, among others, remain unknown. Better understanding of the thermodynamic behavior and phase equilibria could help in developing novel energy- and lifecycle-efficient alloys and processes.
Although ab initio methods have been used to predict intermetallic phase diagrams for a long time, application to real materials in combination with experimental modelling methods (CALPHAD) is a rather new concept. Machine learning methods, such as genetic algorithms and heuristic structure selection, are also applied in this project.
This project has been conducted in collaboration with Prof. Emeritus Pekka Taskinen at the Department of Chemical and Metallurgical Engineering, Aalto University, as well as Prof. Jaakko Akola (Norwegian University of Science and Technology, Tampere University of Technology).
Research highlights:
Catalytic Properties of Magnetic Materials:
Magnetism and magnetic materials have a wide array of applications ranging from medicine to electronics. Quantum mechanics can be used to explain the origins of magnetism in any material, because electrons have an intrinsic magnetic dipole moment. Understanding the magnetic properties of individual atoms allows us to predict what kind of magnetic properties should be prevalent in bulk materials.
The magnetic state of a metal also affects the catalytic properties of the material. By focusing on reactions involving e.g. CO on iron-based nanoclusters and surfaces, our goal has been to quantify these effects and to investigate whether magnetic fields could be used for controlling catalysis. We have also combined the gained understanding of magnetism with our experience in thermodynamic modelling of metal mixtures to study novel alloyed nanoparticles with improved catalytic properties.
A part of this research has been conducted in collaboration with Prof. Hannes Jónsson (University of Iceland, Aalto University).
Research highlights:
Teaching:
The Computational Chemistry group is responsible for arranging graduate (maisteri- ja tohtorivaiheen) level teaching on various lecture and laboratory courses in the Chemistry major of the Master's Programme in Chemical, Biochemical and Materials Engineering. The aim of the lecture courses is to establish the basics of statistical physics, quantum mechanics and quantum chemistry as needed by chemists, as well as to hone previously learned mathematical tools that are required to solve problems in these fields. In the practical hands-on exercise sessions and laboratory assignments, the focus is on giving the students an overview on the sort of systems that can be tackled with the tools of computational chemistry employing software that is in everyday research use in the group. A list of currently taught courses is given below, with additional information available either in Into or in the school syllabus.
- CHEM-E4110 Quantum Mechanics and Spectroscopy (5 cr)
- CHEM-E4115 Computational Chemistry I (5 cr)
- CHEM-E4225 Computational Chemistry II (5 cr)
Our research group:

Head of the research group: Kari Laasonen, Ph.D., Full Professor
Kari Laasonen established the Computational Chemistry research group in 2010, when he was appointed the professor of Physical Chemistry. Before moving to Aalto University, he held the same chair at the University of Oulu between 1996 and 2010.
Kari has a strong background in both physical chemistry and theoretical physics. He received his doctoral degree in theoretical physics at the University of Helsinki, but later he was also awarded the degree of docent in physical chemistry. After graduating, Kari spent many years abroad visiting world-renowned research groups. Notable highlights include Prof. Michele Parrinello's group at the IBM Research Laboratory in Switzerland, and Prof. Michael Klein's group at the University of Pennsylvania in the United States.
Group members:
Michael Busch, Ph.D., Postdoctoral Researcher
Michael received his Ph.D. in chemistry at the University of Gothenburg where he studied water oxidation on transition metal oxides using DFT. He then moved on to do postdoctoral projects at the Technical University of Denmark (DTU), Ecole Polytechnique Fédérale de Lausanne (EPFL) and Chalmers University of Technology. After complementing his academic skills by working at the R&D department of the industrial company Nouryon, he joined the Computational Chemistry group at Aalto University where he continues modelling electrochemical and photochemical reactions with relation to fuel cells and electrolysis.
Arsalan Hashemi, D.Sc. (Tech.), Postdoctoral Researcher
Aliya Mehranfar, Ph.D., Postdoctoral Researcher
Rasmus Kronberg, M.Sc. (Tech.), Doctoral Candidate
Rasmus completed his M.Sc. degree in 2018 in the Computational Chemistry group. The title of his thesis was "Ab initio molecular dynamics simulation of the platinum–water interface: Insights into structure and electrocatalytic properties". In his doctoral thesis, Rasmus continues modelling electrochemical interfaces and reactions using dynamic DFT-based methods.
Mario Mäkinen, M.Sc. (Tech.), Doctoral Candidate
Mario finished his M.Sc. degree in 2019 in the Computational Chemistry group. His thesis regarding atomic and molecular layer deposition (ALD/MLD) had the title "Density functional theory based modelling of hybrid zinc oxide thin films". In his doctoral thesis project, Mario continues to model functional surface structures and surface phenomena.
Reza Khakpour, M.Sc., Doctoral Candidate
Reza obtained his master's degree in nanotechnology at the Kavosh Institute of Higher Education, Iran. His thesis was titled "A new durable and high-performance platinum supported on Ag-Ni-porous coordination polymer as an anodic DMFC nano-electrocatalyst: DFT and experimental investigation". As a doctoral candidate at the Computational Chemistry group, Reza will focus on DFT modelling of new catalyst materials for the electrochemical reduction of CO2.
Heikki Lappalainen, B.Sc. (Tech.), Master's Thesis Worker
Heikki is a master's thesis worker in the group of Computational Chemistry. In his thesis project, Heikki explores reaction databases and machine learning algorithms for activation energy prediction in the spirit of data-driven catalyst design.
Postdoctoral researchers and Research Fellows:
Matthias Vandichel, Geraldine Cilpa-Karhu, Garold Murdachaew, Lauri Partanen, Mikko Hakala, Richard Hatz, Jian-Cheng Chen, Olga Syzgantseva, Suvi Ikäläinen, Divya Srivastava, Stefan Taubert
Doctoral Candidates:
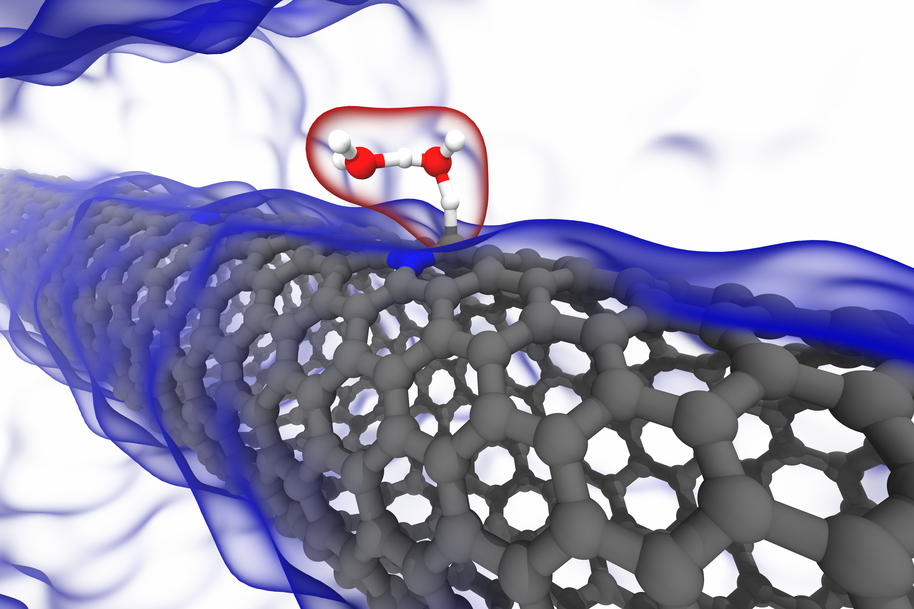
Nico Holmberg, D.Sc. thesis title "Hydrogen Evolution Reaction on Carbon Nanotubes: Insights from Electronic Structure Theory"
Timo Weckman, D.Sc. thesis title "First Principles Multiscale Modelling of the Atomic Layer Deposition of Al2O3 and ZnO"
Olli Pakkanen, Lis.Sc. thesis title "Towards Cleaner Energy with Carbon-Based Catalysts: Density Functional Theory Studies of the Hydrogen Evolution Reaction"
Juhani Teeriniemi, D.Sc. thesis title "Thermodynamic Modeling of Alloys by Coarse-grained Density Functional Theory"
Marko Melander, D.Sc. thesis title "Reactivity of Iron Nanostructures from Density Functional Theory"
Undergraduate and M.Sc. thesis students
Katja Kohopää, Mikael Särkkä, Ilona Pohjavirta, Saana Lipasti, Tarmo Nurmi, Jari Huisman, Sanna Renkonen, Emilia Stenman, Eelis Solala, Ville Latsa
Job openings:
- For the current status of job openings including M.Sc. thesis positions related to currently active research projects, please contact Prof. Laasonen. Doctoral student candidates may apply at M.Sc. thesis level. B.Sc. thesis topics in physical chemistry with a focus on computational chemistry are available upon request.
- We hire annually 1-3 undergraduate (B.Sc. or M.Sc.) students as summer research assistants. The application period is in January.
- All applications with the appropriate attachments (e.g. curriculum vitae) should be sent to the research group leader, unless otherwise stated.
Related content:
Put the brakes on climate change – producing high-grade chemicals from carbon dioxide
A study aiming to use CO2 as, for example, transport fuel have received significant funding from the Jane and Aatos Erkko Foundation.

Promising results obtained with a new electrocatalyst that reduces the need for platinum
Researchers succeeded in manufacturing electrocatalysts with one hundredth of the amount of platinum that is usually needed.
Critical catalyst materials research receives funding from Horizon 2020
The research aims to provide alternatives to critical metals used in certain chemical reactions and energy conversions.
Researchers developed a cost-effective and efficient rival for platinum
Researchers succeeded in creating an electrocatalyst that is needed for storing electric energy made of carbon and iron.
Latest publications:
DFT Mechanistic Investigation into Ni(II)-Catalyzed Hydroxylation of Benzene to Phenol by H2O2
Kinetics of N2 Release from Diazo Compounds: A Combined Machine Learning-Density Functional Theory Study
Divergent Mechanistic Scenarios for the C−sp2 and C−sp3 C−H Insertion of Aurated Aryl Cations
Density functional theory and machine learning for electrochemical square-scheme prediction: an application to quinone-type molecules relevant to redox flow batteries
Understanding Electron Transfer Reactions Using Constrained Density Functional Theory: Complications Due to Surface Interactions
Temperature-Controlled Syngas Production via Electrochemical CO2 Reduction on a CoTPP/MWCNT Composite in a Flow Cell
CO2 or Carbonates – What is the Active Species in Electrochemical CO2 Reduction over Fe-Porphyrin?
Selectivity of CO2, carbonic acid and bicarbonate electroreduction over Iron-porphyrin catalyst: A DFT study
Density functional theory study of trends in water dissociation on oxygen-preadsorbed and pure transition metal surfaces
An unexpected synthesis of azepinone derivatives through a metal-free photochemical cascade reaction
- Published:
- Updated: